
1 min read
Kepler Microbiome Analysis: Tools & Applications
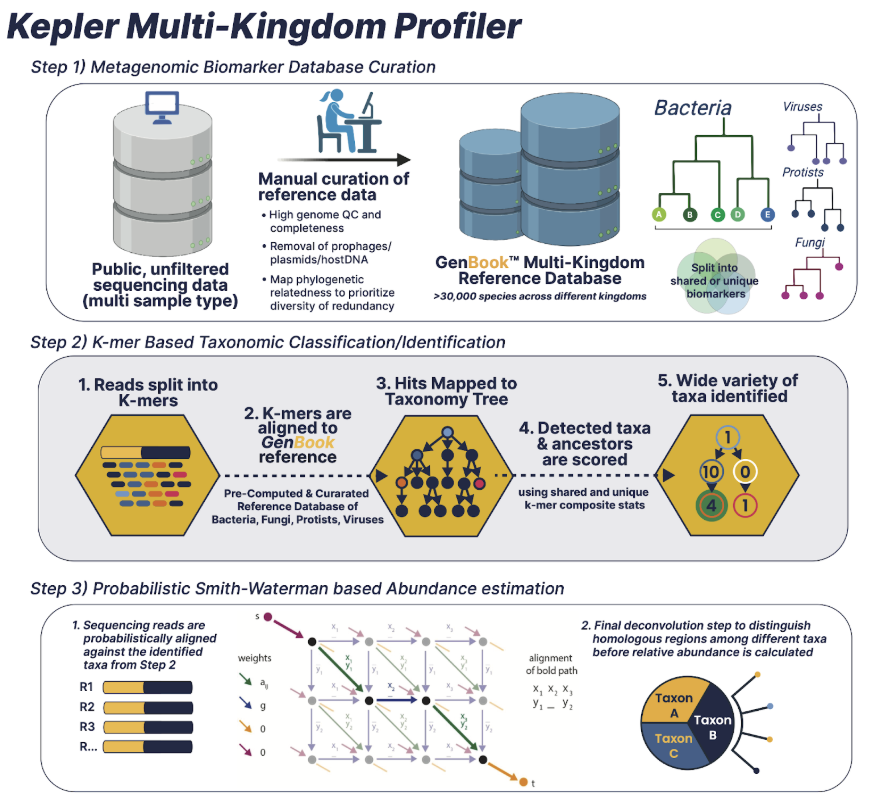
The Kepler metagenomic profiler is a host-agnostic pipeline focused on taxonomic and functional profiling. Powered by Cosmos-Hub, Kepler is...
4 min read

EMU is a computational pipeline built for high-accuracy species-level profiling in microbial communities using full-length 16S rRNA gene reads. The platform is uniquely optimized for long-read sequencing technologies such as Oxford Nanopore and PacBio (read more about our approach in the Microbiome Profiling section).
By implementing an expectation-maximization algorithm, EMU excels at correcting sequencing error and refining taxonomic classification with every iteration, supporting researchers who require accurate, full-length 16S analyses and species-level abundance estimates.
EMU demonstrates improved species-level profiling accuracy and reduced false positives in comparison to alternative methods. (Curry et al., 2022, Nat Methods).
EMU pipeline directly addresses the challenge of species-level resolution in amplicon-based microbiome analysis. Unlike short-read amplicon assays that restrict taxonomic assignments mostly to the genus level, EMU characterizes diversity across closely related microbes by analyzing the complete ~1,550bp 16S rRNA gene (compared to ~250bp fragment captured with short-read 16S). This additional coverage leads to a more detailed overview of the 16S genes present in the sample, providing species-level taxonomic abundance outputs for any sample type, particularly low-biomass and high-host content (i.e. tissue). .
Researchers can also interpret taxonomic abundance with improved clarity, thanks to EMU’s sophisticated error model and minimap2-based alignments providing accurate taxonomy lineages, even from error-rich Oxford Nanopore Technologies reads.
Learn more about species-level profiling capabilities and datasets in the Content Library.
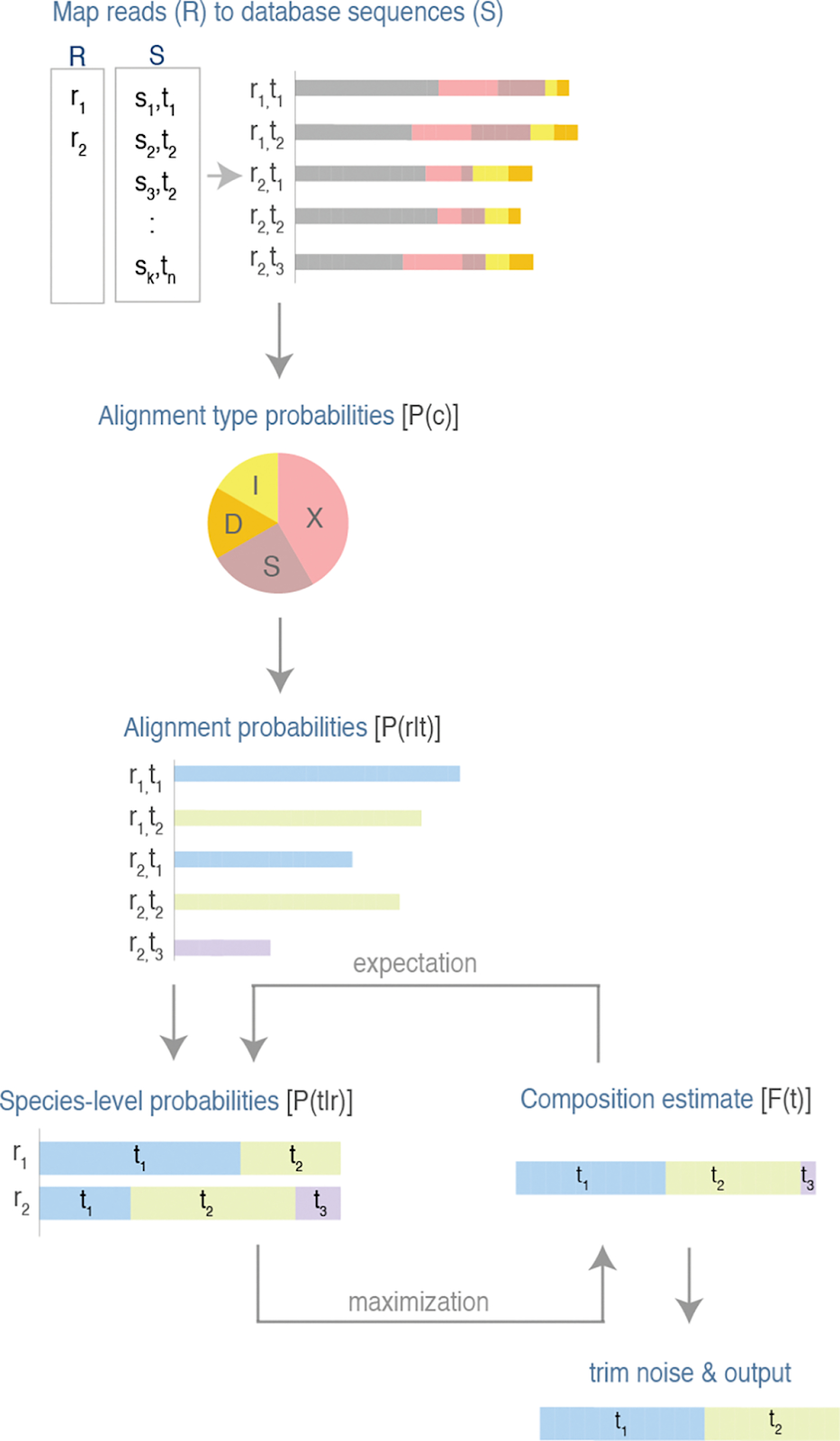
Figure 1. Pictorial representation of Emu algorithm
By enabling detailed profiling, EMU allows researchers to detect species that remain invisible to other platforms, enhancing species detection in clinical studies, ecological monitoring, and food safety research.
EMU delivers iterative, error-aware taxonomic profiling. Core features of the EMU analytical workflow include:
For advanced benchmarking and throughput, the Cosmos-Hub platform offers a number of methodologies via the Statistics Toolbox.
EMU outperforms alternative tools in species-level resolution and error minimization.
EMU supports extensive curatedreference databases.
Adaptable database selection ensures EMU remains relevant as microbial taxonomy evolves, supporting innovation and routine application alike.
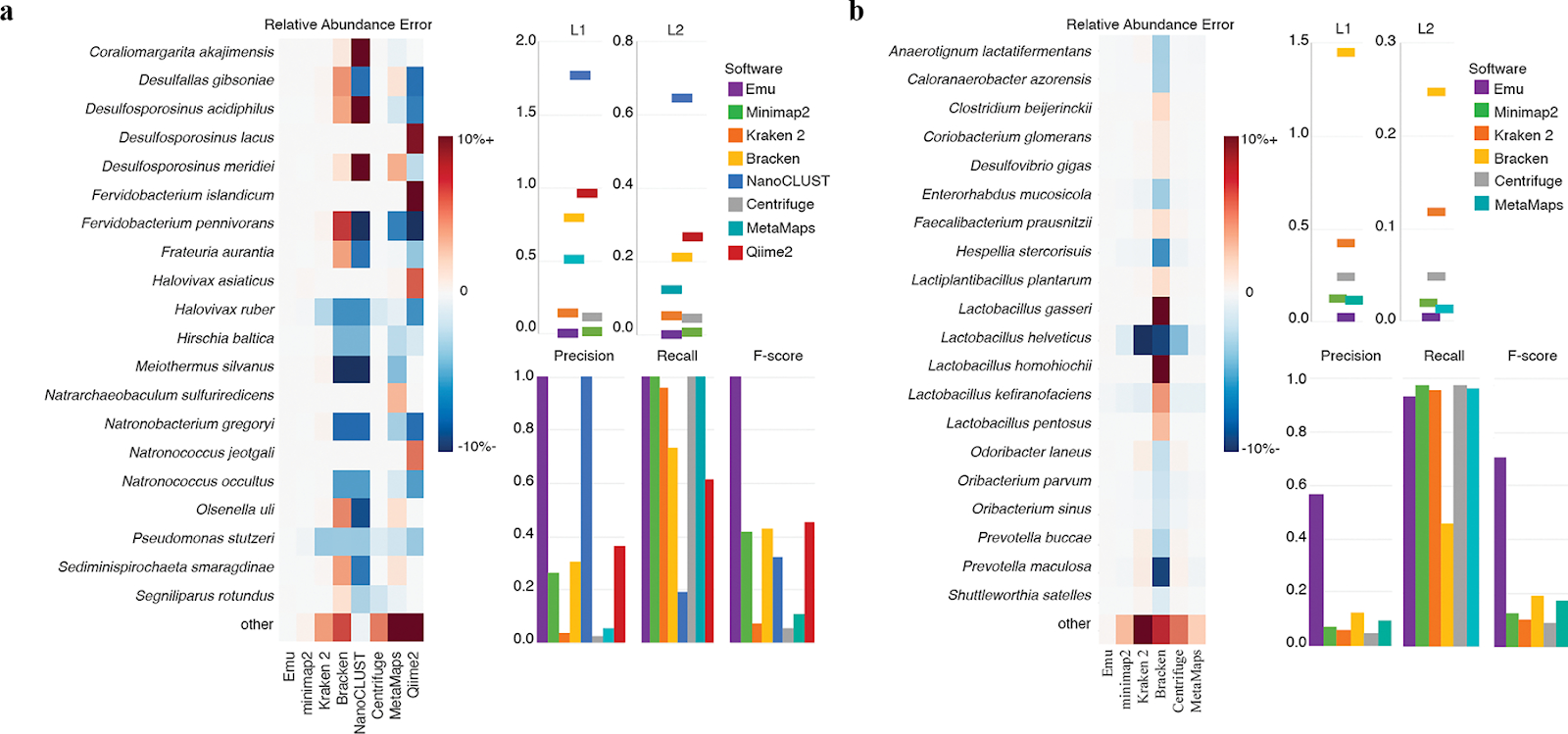
Figure 2. Performance on simulated ONT reads via EMU Pipelines
EMU can be easily run with customized parameters in the Cosmos-Hub web-based GUI, compared to traditional command-line options. Additionally, we have optimized EMU’s performance along with integrated multiple databases, enabling researchers to quickly profile their data and create comparative analyses.
The only requirements for running EMU via the Cosmos-Hub are high-quality FASTQ files produced by a long-read amplicon workflow (16S, ITS, 18S) and a web-browser. No other computational requirements!
By default, EMU restricts ultra-low-abundance taxa (species below 1–10 reads), enabling a focus on robust, interpretable results. Users interested in examining only a portion of their sample or needing to detect every possible community member can change abundance output thresholds or supplement with alternative data processing approaches.
Key challenges to plan ahead of include:
Nonetheless, EMU offers the most effective model for robust, reproducible species-level microbiome studies in mainstream applications.
Setting up EMU locally can be a technical headache. Researchers must install Conda, configure Python ≥3.6, manage databases from various sources, and optimize minimap2 to handle large datasets without memory issues.
Multi-threading, batch sizes, and output structures all require careful tuning for high-throughput analysis, and integration with R, Snakemake, or cloud workflows adds another layer of complexity. Even after setup, interpreting results demands attention to abundance thresholds and handling ultra-low-abundance taxa, which can be particularly tricky in diverse microbial communities.
Cosmos-Hub eliminates these barriers by delivering powerful, no-code access to EMU pipelines. With Cosmos-Hub:
Whether you’re a lab scientist, bioinformatician, or industry professional, Cosmos-Hub removes the technical friction so you can focus on what matters most: interpreting microbiome data and generating actionable insights.
EMU specializes in full-length, species-level profiling and incorporates iterative error correction, reducing false positives compared to genus-level or short-read platforms.
EMU is validated on gut, vaginal, marine, soil, synthetic, environmental, and clinical samples, demonstrating versatility and reliability across research contexts.
EMU’s EM algorithm is specifically configured for full-length 16S rRNA profiling, offering a tailored implementation not commonly found in other platforms. Cmbio has also added additional optimizations to ensure seamless performance, which is discussed further in our documentation (https://docs.cosmosid.com/docs/emu).
1 min read
The Kepler metagenomic profiler is a host-agnostic pipeline focused on taxonomic and functional profiling. Powered by Cosmos-Hub, Kepler is...
1 min read
In the realm of microbiology, accurately identifying bacterial species is crucial for understanding microbial communities, diagnosing infections, and...
1 min read
DADA2 is an open-source R package for accurate sample inference on amplicon sequencing data, outputting fewer spurious sequences while generating...