
1 min read
Bioinformatics Pipelines in Microbiome Analysis: A Comprehensive Guide
A bioinformatics pipeline is the central engine driving microbiome analysis, turning raw sequencing data into interpretable results. Every aspect of...
4 min read
DADA2 is an open-source R package for accurate sample inference on amplicon sequencing data, outputting fewer spurious sequences while generating exact amplicon sequence variants (ASVs) from paired-end fastq files. The DADA2 pipeline leverages a sophisticated, data-driven error model using estimated error rates from the user's own data, delivering high resolution sample inference and enabling researchers to resolve biological differences down to single nucleotide changes.
In this article, we explore the core innovations that underlie DADA2, dissect its stepwise pipeline for high-resolution ASV determination, and explain how its approach overcomes key limitations of OTU clustering. We’ll compare DADA2’s data-driven error correction, strain-level variant calling, and universal feature labeling with legacy methods, clarifying why DADA2 now sets the benchmark for reproducible, scalable microbiome research.
The DADA2 software was developed by leading figures in microbiome data science, primarily Benjamin J. Callahan, alongside Paul McMurdie and Susan Holmes, with substantial contributions recognized throughout the project’s history and in its rigorous benchmarking publications. It is maintained and actively developed under an LGPL license, and supported by an open, collaborative community hosted on Bioconductor and GitHub.
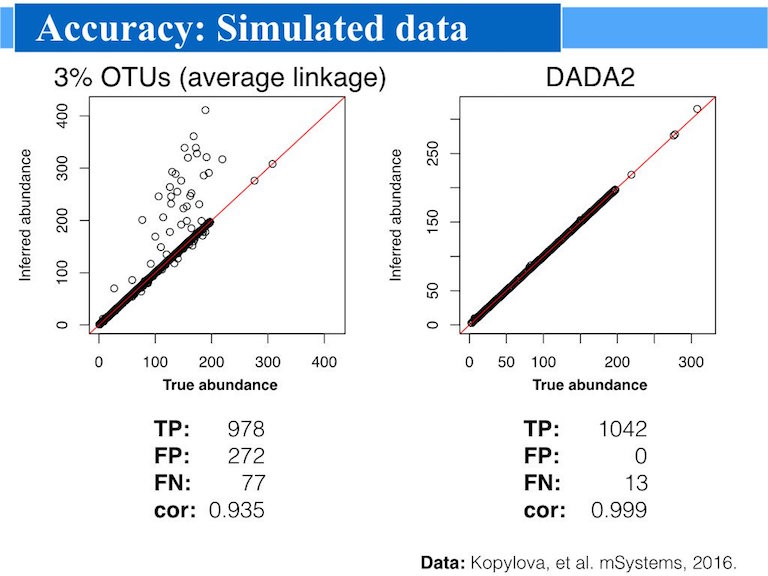
DADA2 infers exact amplicon sequence variants—supporting workflows that require precise sequence variants for further analysis and universal reference sequences for meta-analysis. Unlike traditional OTU-based methods (e.g., QIIME2, mothur), which group output sequences using 97% identity clustering,
DADA2 uses its error model to identify unique sequence variants inferred within the sequencing data itself .This capability allows for the identification of real biological variation in microbiome studies that OTU clustering would miss or obscure.
|
Feature |
DADA2 |
OTU-based Methods |
|
Resolution |
Single nucleotide |
97% identity clustering |
|
Error model usage |
Data-driven |
Generic or none |
|
Output type |
ASVs |
OTUs |
|
Cross-study comparability |
Direct |
Only by re-clustering |
|
Spurious results |
Low |
Higher |
This innovation empowers researchers to probe microbial diversity at a much finer scale, capturing subtle variants invisible to traditional pipelines.
DADA2’s amplicon sequencing workflows apply error correction and sample inference at each stage, ensuring reliable identification of sequence variants. The major steps, each informed by quality score and trimming parameters, include:
Each processing stage is crucial: error modeling is central to DADA2’s accuracy, while chimera removal safeguards the validity of downstream analyses.
DADA2 delivers precision, accuracy, and scalability. Its design and output empower microbiome researchers with tools for robust, high-resolution ecological studies.
Because ASVs are universally comparable, meta-analyses and large consortium data integration become straightforward.
Cosmos-Hub is a no-code, cloud-based microbiome analysis platform that integrates DADA2 and other bioinformatics pipelines to streamline microbiome profiling. It offers several features to address the limitations associated with DADA2:
By integrating these features, Cosmos-Hub enhances the usability and effectiveness of DADA2, addressing common challenges in microbiome data analysis.
Access DADA2 and related tools in Cosmos-Hub—a web-based, no-code microbiome analysis platform integrating reference sequences, sample composition data, and advanced amplicon sequencing workflows for all researchers. For a tailored demo or enterprise solution, click below:
By modeling errors and distinguishing true variants at the single-nucleotide level, DADA2 enables the study of microbial populations with finer granularity than OTU clustering.
DADA2 is designed for use with Amplicon sequencing data produced by short-read sequencing technologies (for long-read amplicon data, the EMU pipeline is available). DADA2 generates a precise sequence table that catalogs the exact biological sequence variants identified across samples in amplicon sequencing data.
DADA2 works best with demultiplexed, high-quality, paired-end FASTQ files from platforms like Illumina and Element. It can be adapted for varied amplicons, including bacterial 16S and fungal ITS regions, although specific workflows and modifications may be needed for certain marker genes.
As the ITS sequence is a bit more heterogeneous across species, we support OTU profiling of ITS data with QIIME2 and the UNITE database. The workflow requires additional steps, especially for primer removal, and accounts for length variability characteristic of ITS amplicons.

1 min read
A bioinformatics pipeline is the central engine driving microbiome analysis, turning raw sequencing data into interpretable results. Every aspect of...
1 min read
In the realm of microbiology, understanding the intricate workings of microbial communities has become a cornerstone of scientific inquiry. Two...

1 min read
Background